FAQ: Medical Device Regulation (MDR)
1. how can I recognise whether a medical device is MDR compliant?
Conformity can still be recognised by the CE mark on the type plate. The "MD" for "Medical Device" is new. The product itself has not changed. You will also receive new operating instructions and a new declaration of conformity for the product.
You can find the current operating instructions in the download area on our website https://www.bock.net/unternehmen/downloads/
Your Bock contact will be happy to provide you with the new declaration of conformity.
Example of type plate:
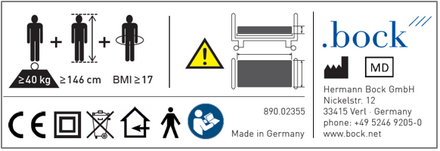
3. what does making available on the market ("sale of the MP") under the MDR mean for me as a distributor?
According to the MDR, you as the dealer make the nursing care bed available for the market until the time of its commissioning. This entails a number of tasks and obligations for you. For example, checking that the product bears the required CE marking, that an EU declaration of conformity (issued by Bock as the manufacturer) is available and, in particular, that the instructions for use are enclosed with each product. In the future, the verification of the UDI will also become relevant (see question 6).
In addition, the MDR now explicitly stipulates that you as a distributor must ensure that the storage and transport conditions that we at Bock provide for the products are also complied with on site or by you.
Market observation is also a new and important point in the MDR, for example to improve products based on user feedback. In addition, Bock wants to and must ensure good communication and cooperation between manufacturers and distributors and, if necessary, the competent authorities.
If you as a distributor ever receive complaints and reports from healthcare professionals, patients or users about suspected incidents, please forward them to us immediately so that we can take further action if necessary. Reports to the BfArM by you as a distributor may also be relevant if you determine that a product poses a serious risk.
Incidents can be, for example, application errors, problems with operation, malfunctions or other feedback regarding the properties or performance of the products.
In this context, we would like to point out that, as a distributor, you must keep a register in accordance with the MDR in which you document possible complaints, information on conformity problems, any recalls and withdrawals. You must keep Bock up to date with this register.
4. what are the most important requirements for bock?
As a manufacturer of Class I medical devices, bock has extensive tasks and obligations to fulfil according to the MDR and must of course ensure that you receive safe medical devices from our company. The most important requirements for Bock are the creation and maintenance of technical documentation, proof of EU conformity, risk management and clinical evaluation. As a manufacturer, we must of course also affix the CE mark to the products. If you do experience any problems or actually notice any, please let us know immediately, as required by law.
As already mentioned, the MDR has also increased the importance of active market surveillance, which Bock would like to address and ensure together with you and your information (see question 3).
5 What impact does the MDR have on liability?
In terms of liability, nothing has changed for bock as the manufacturer - at least nothing new has been added. However, you as a dealer have been given more tasks to check.
If you do not fulfil these obligations, this may in turn have consequences under liability law. You are also still liable to an injured party to the same extent. Although these liability claims can be passed on to Bock as the manufacturer, you must first have sufficient financial security yourself.
In connection with a possible liability, a possible claim for damages by natural or legal persons is always possible, also in accordance with the MDR, and must be financially secured accordingly.
6 What is UDI, what does it mean for me as a retailer and for the traceability of products?
The abbreviation UDI stands for Unique Device Identifier - UDI.
The new UDI system is used to identify and register medical devices and is intended to enable fast and secure traceability along the entire supply chain:
Identify medical devices more easily
Trace locations quickly and easily
Find and detect illegal medical devices more quickly
This should enable quick and efficient recalls in the event of safety risks and facilitate the necessary dialogue with the relevant authorities.
The principle is very simple: each manufacturer receives unique identification numbers for each medical device from an authorisation body, which in turn must be clearly visible on the product and packaging.
The MDR distinguishes between the basic UDI-DI, the UDI-DI and the UDI-PI:
Basic UDI-DI:
An identification number for a group / several variants of a manufacturer's medical device
Primary identifier of a device model, at the level of the unit of use
Most important organisational feature
For example, the basic UDI is specified in the declaration of conformity.
Bock's basic UDI-DI: 4063588HERMANNBOCKRH
UDI-DI:
The device identification of a specific model (e.g. the domiflex bed)
Serves as a key in the UDI database
Each UDI-DI belongs to exactly one basic UDI-DI
Bock's UDI-DI: 40635880000
UDI-PI:
The production identifier identifies each individual instance of a product
At Bock, each nursing care bed is thus assigned a unique serial number by the UDI-PI
You will find the UDI-DI and UDI-PI as a code on the type plate on the bed itself and in the declaration of conformity.
The only obligation for you as a dealer is to check the products and ensure that the type plate remains on the nursing care bed at all times.
The EU Commission is setting up EUDAMED as a central database for medical devices. This is expected to go live in May 2022 and will be publicly accessible free of charge.
IMPORTANT: The UDI does not have to be affixed to Class I medical devices until 26 May 2025.
7 What is the difference between making available and placing on the market?
Placing on the market" refers to the first time a product is made available on the Union market. For example, when the manufacturer delivers a product to the retailer or when an importer imports a product into the European Union for the first time. Placing on the market is therefore the starting point of the supply chain in the Union.
Placing on the market is followed by "making available on the market". The product is offered for sale or use in the course of a commercial activity, whether in return for payment or free of charge. This is the case, for example, when a medical supply retailer offers a product to a customer.
As soon as a ready-to-use product is available to the end user and the end user can use the product in accordance with the intended purpose defined by the manufacturer, the time of "putting into service" is reached.
8 What happens next?
In addition to the requirements of the European MDR, you as a distributor may also be subject to obligations arising from national regulations that only apply in Germany. For example, the Medical Devices Operator Ordinance (MPBetreibV) regulates both general and specific requirements for the installation, operation, use and maintenance of medical devices. The new Medical Device User Notification and Information Ordinance (MPAMIV) also regulates the reporting of incidents. It must therefore be observed by you, particularly in connection with the obligations explained under question 3.
The national regulations focus on the actual use and handling of the products, whereas the MDR focuses primarily on the manufacture, placing on the market and monitoring of medical devices.
As a distributor, you may also fall under the operator obligations if statutory care and health insurance funds delegate the tasks (see Section 3 MPBetreibV) to you as a service provider by contract in accordance with Section 126 SGB V. However, this may also be the case with other cost bearers such as private health insurance companies or accident insurers.
Please note the respective national regulations and special features outside Germany.
9 What are the most important requirements for the specialised trade / operator?
Both European and national regulations oblige retailers and operators to fulfil certain tasks.
For example, a dealer must check the requirements for the provision of a product before making it available (see question 7).
A distributor must also observe the manufacturer's specifications, for example on technical safety during assembly/installation (so-called "setting up" of the product), but also on hygiene, contamination, cleanliness, infection protection and reprocessing as well as transport and storage conditions
A distributor also assumes responsibility for product selection and control, taking into account the intended purpose defined by the manufacturer.
In addition, the retailer must develop and maintain a traceability system together with the manufacturer and fulfil the reporting obligations, i.e. he must report incidents in compliance with the reporting deadlines and record complaints and pass them on to the manufacturer.
If you are also the operator (see question 8), you are organisationally responsible in particular for the following activities in connection with the respective medical device. This means that you must ensure that these are actually carried out, either by yourself or by the users:
setting up, i.e. assembling, assembling, installing, etc.,
the provision
Maintenance, which consists of repairs (servicing, repairs, etc.) and other maintenance measures,
reconditioning and
safety and metrological checks
the use of the products in accordance with their intended purpose
Various documentation obligations, in particular the maintenance of an inventory, must also be observed. Please note that there are no safety checks for nursing care beds, only technical checks, the intervals of which are to be determined by the operator.
Culpable violations by the operator, including negligent behaviour, can lead to liability in accordance with § 823 BGB.
Please obtain comprehensive information on these special obligations, as operators must ensure that medical devices in their area of responsibility are used with due care and are not faulty. They therefore have a duty of care.
Outside Germany, please observe the respective national regulations and special features.
If you have further questions about the MDR or need detailed information, please send us a message to or visit the following websites:
Daily updated information on the MDR as a database: https://lexparency.de/eu/MDR/
MDR page of the EU Commission: : https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX:02017R0745-20200424
Please also note the respective national regulations and special features.